Variance propagation diagnostic for rarefaction
Source:R/plot_variance_propagation.R
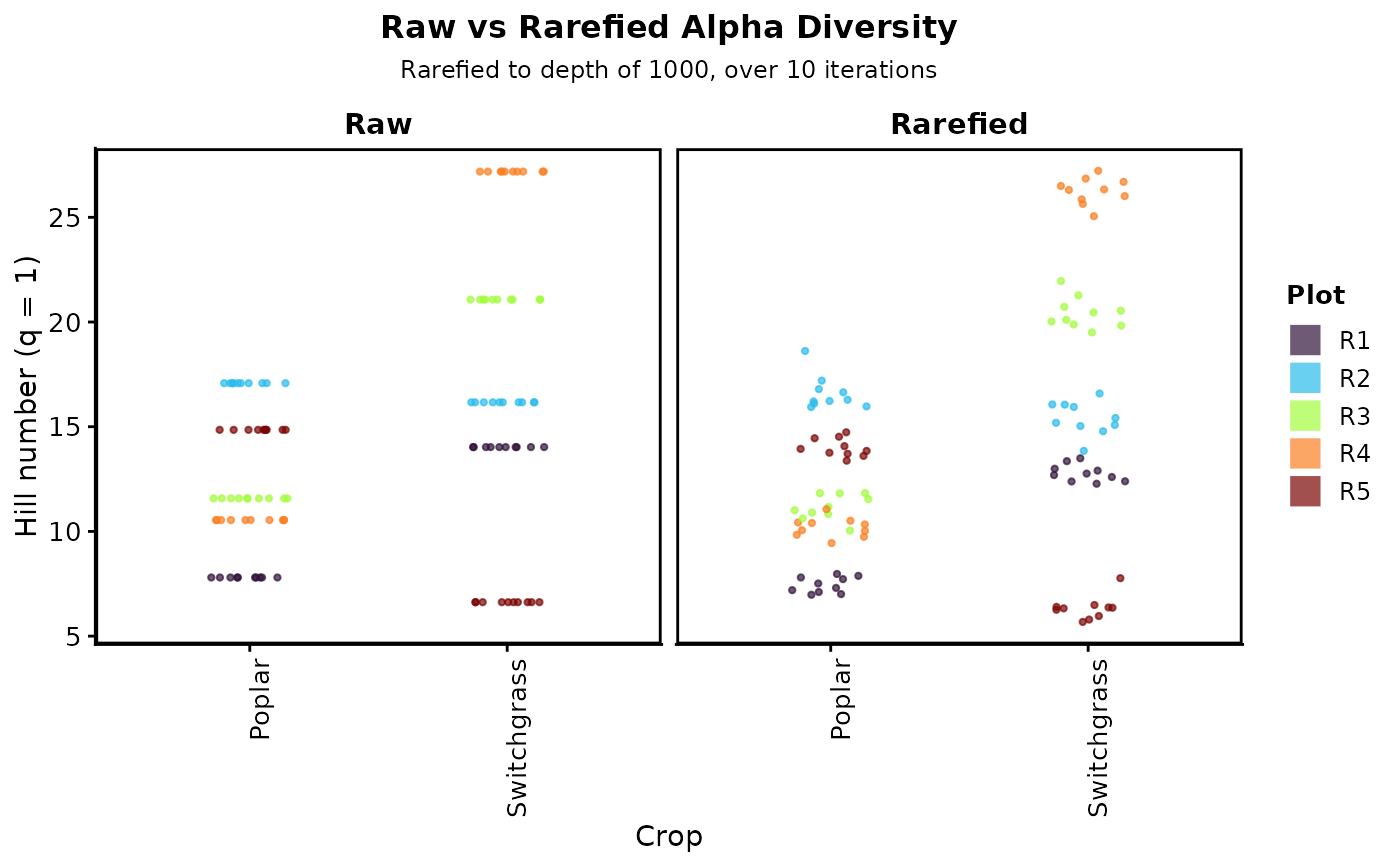
plot_variance_propagation.RdThis function evaluate the variance between rarefaction iterations from multi_rarefy() by visually comparing raw vs. rarefied alpha diversity metrics calculated at each iterations. It is possible to plot observed richness (q=0), Shannon diversity (q=1), or Simpson diversity (q=2) by setting the q parameter to "richness" or q = 0, "shannon" or q = 1, or "shannon" or q = 2. The plot is faceted by method (raw vs rarefied) and colored by a specified grouping variable from the sample data.
Usage
plot_variance_propagation(
physeq_obj,
rarefied,
q = 0,
group_var,
group_color,
convert_to_factor = FALSE
)Arguments
- physeq_obj
Raw phyloseq object
- rarefied
Output from multi_rarefy(). Either a list of dataframes or and array.
- q
Hill number order (q = 0 for richness, q = 1 for Shannon, q = 2 for Simpson)
- group_var
A grouping variable to use gor grouping as in the sample_data()
- group_color
A color variable to use present in the sample_data()
- convert_to_factor
Logical. If
TRUE, bothgroup_varandgroup_colorare coerced tofactorbefore plotting, which is useful when those columns are numeric/continuous (e.g. dates, counts) but should be treated as discrete groups. WhenTRUEa discrete color scale (scale_color_viridis_d) is used; otherwise the continuous scale (scale_color_viridis_c) is used. DefaultFALSE.
Examples
library(phyloseq)
library(BRCore)
# Example comparing hill q=1 between Poplar and Switchgrass plots
bcse_filt <- bcse |>
subset_samples(Crop %in% c("Poplar", "Switchgrass"))
bcse_rarefied_otutable_filt <-
multi_rarefy(
physeq_obj = bcse_filt,
depth_level = 1000,
num_iter = 10,
.as = "list",
set_seed = 7643
)
#>
#> ── Rarefaction iterations starting... ──────────────────────────────────────────
#>
#> ── Input Validation ──
#>
#> ✔ Input phyloseq object is valid!
#> ℹ Seed: 7643
#> ℹ Input (matrix/df dim): 10 samples x 2861 taxa
#> ℹ Rarefaction depth: 1000
#> ℹ Iterations: 10
#> ℹ taxa_are_rows: TRUE
#> ℹ OTU matrix/df rownames head: bcse73, bcse102, bcse104, bcse77, bcse78, bcse75
#> ℹ OTU matrix/df colnames head: OTU_427, OTU_11, OTU_253, OTU_148, OTU_3, OTU_78
#> ℹ Row sums summary: Min=3146, Max=67815, Median=8685.5
#>
#> ── Rarefaction Results ──
#>
#> ── Sample Removal
#> ✔ No samples removed.
#>
#> ── Taxa Removal
#> ✔ No taxa removed.
#> ! Taxa are not removed across iterations to maintain consistent dimensions.
#> Downstream analyses should handle zero-abundance taxa appropriately.
#>
#> ── Data Sparsity
#> ℹ Returning list of data frames for each iteration.
#> • Rarefied matrix (across 10 iterations):
#> • Min: 27998 zeros (97.86% sparsity) out of 28610 entries
#> • Max: 28029 zeros (97.97% sparsity) out of 28610 entries
#> • Avg: 28011.6 zeros (97.91% sparsity) out of 28610 entries
#>
#> ── Final Data Dimensions
#> ✔ Output: 10 iterations with 10 unique samples
#> • Samples per iteration:
#> • Min: 10
#> • Max: 10
#> • Non-zero taxa per iteration:
#> • Min: 188
#> • Max: 200
#> • Avg: 193.1
plot_variance_propagation(
physeq_obj = bcse_filt,
rarefied = bcse_rarefied_otutable_filt,
q = 1,
group_var = "Crop",
group_color = "Plot"
)
#> ✔ Input phyloseq object is valid!
#>
#> ── Rarefaction Variance Propagation Visualization ──────────────────────────────
#> ℹ Hill number order selected, q= 1
#> ℹ Number of rarefaction iterations, n_iter= 10
#> ℹ Comparison plot generated!